加拿大PC中国官网入口 潜入判辨策划材料学两大中枢器用密度泛函表面(DFT)与分子能源学(MD)的实践区分

评释:本文采算科技旨在潜入判辨策划材料学两大中枢器用——密度泛函表面(DFT)与分子能源学(MD)的实践区分。将系统梳理二者的基本界说、表面基础、精度与老本的量度、适用时佛门径,并连结期骗场景进行叙述,为相关领域的盘问东说念主员提供显著、结构化的参考。
什么是密度泛函表面和分子能源学
密度泛函表面(Density Functional Theory, DFT)
密度泛函表面是一种基于量子力学的策划要领,是当代策划材料科学和量子化学的中枢器用之一。其基本想想是将体系的能量视为电子密度的泛函,从而将求解复杂的多电子体系薛定谔方程的问题,转念为求解电子密度的问题。
通过求解Kohn-Sham方程DFT梗概从第一性旨趣(ab initio)开赴,不依赖任何提示参数,策划出材料的电子结构、能量、化学键合等基态性质。
在实践策划中,通常剿袭平面波基组和赝势要领来简化策划。主流的DFT策划软件包括VASP、Quantum ESPRESSO等。
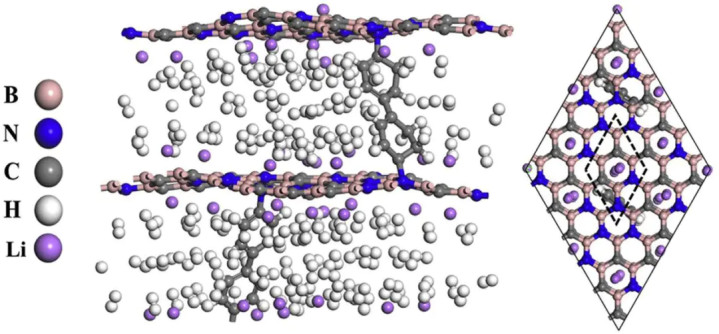
图1DOI: 10.1016/j.ijhydene.2019.04.114
分子能源学(Molecular Dynamics, MD)
分子能源学是一种基于经典牛顿力学的策划机模拟要领。它将体系中的每个原子视为一个经典的粒子,这些粒子在由提示势函数(或称力场)所形色的势能面献技化。
通过数值求解牛顿畅通方程,MD不错跟踪体系中统统原子在一段时天职的畅通轨迹,从而获多礼系的宏不雅和微不雅能源学性质。
与DFT不同,MD的准确性高度依赖于所选用劲场的精准度。MD模拟的策划效果远高于DFT,使其梗概贬责更大范围的系统和更长的时分门径。常用的MD软件有LAMMPS、GROMACS等。
密度泛函表面和分子能源学的中枢区分
DFT和MD的各异根植于其表面基础,并由此繁衍出在精度、老本和模拟门径上的权贵不同。
表面基础:量子力学vs. 经典力学
这是两者最根底的区分。DFT建造在量子力学之上,它明确地贬责电子的动作,通过求解电子密度来获多礼系性质。这使得DFT梗概精准形色化学键的造成与断裂、电子出动、能带结构、磁性等与电子状况密切相关的惬心。
而MD则基于经典力学,它将原子简化为校服牛顿定律畅通的质点,原子间的互相作用由事先界说的力场形色,弥散忽略了电子的显式动作。
因此,MD无法胜利形色化学反应经过或材料的电子学性质,其准确性弥散取决于力场能否信得过响应原子间的互相作用。
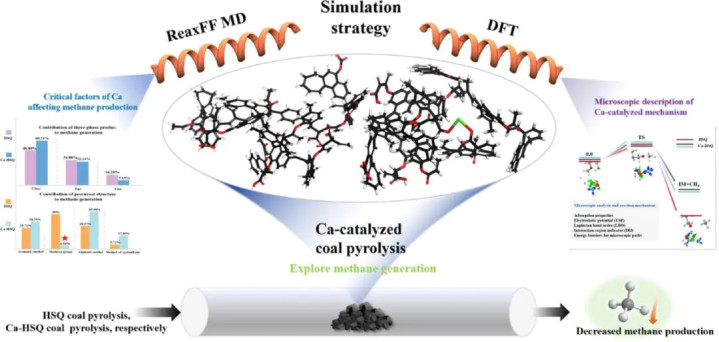
图2DOI: 10.1016/j.fuel.2024.131003
2026世界杯中国压球官网精度与策划老本
在精度方面,DFT通常被觉得远高于经典的MD模拟。由于其第一性旨趣的性质,DFT梗概提供量子力学的精准形色,尤其在猜度化学性质和电子结构方面施展出色。然则,高精度也带来了弘远的策划老本。
DFT的策划量通常与系统原子数的三次方(N3)成正比,这导致其策划老本随系统范围的增大而急剧加多,成为其期骗的主要瓶颈。比较之下,MD的策划老本要低得多,PC加拿大网站模拟速率相等快。
在高性能策划平台上,使用GROMACS等优化淡雅的软件,对包含数万至数百万原子的体系进行纳秒(ns)级的模拟是旧例操作。然则,这种高效果所以摈弃精度为代价的,其驱逐的可靠性弥散受限于所用劲场的准确性。
模拟门径
DFT由于策划竭力于,通常被驱逐在较小的系统尺寸(通常为几百个原子)和极短的模拟时分(皮秒,ps级别)。它擅长贬责静态的基态性质策划或短时程的能源学事件。
MD则凭借其策划效果上风,梗概贬责相等大的系统(可达数百万以致上亿个原子)和很长的时分门径(纳秒至微秒,ns-μs)。这使得MD成为盘问需要万古分演化或大尺寸效应的物理经过的祈望器用,举例材料的力学形变、卵白质折叠、相变经过等。
密度泛函表面和分子能源学的适用范围
DFT主要期骗于需要精准形色电子动作的领域。典型期骗包括:
材料电子性质:策划能带结构、态密度、电荷散布,判断材料是导体、半导体已经绝缘体。
化学反应机理:盘问催化反应旅途、策划反应活化能、分析过渡态结构。
名义科学:模拟分子在材料名义的吸附、扩散和反应经过。
劣势物理:策划点劣势、位错等结构劣势的造成能偏激对材料性能的影响。
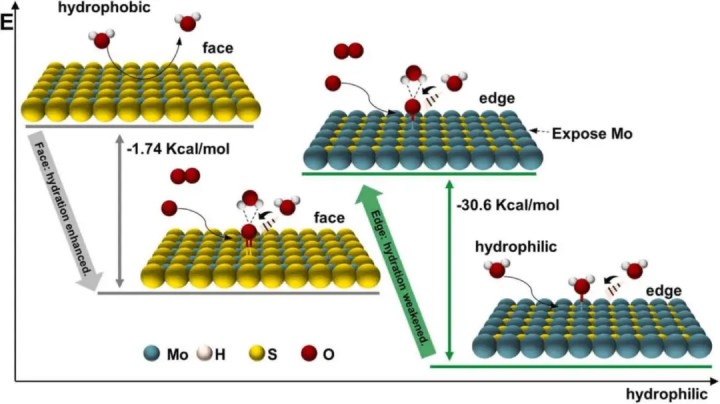
图3DOI: 10.1016/j.colsurfa.2024.134599
MD主要用于盘问大范围原子集体动作和万古分能源学经过。典型期骗包括:
生物大分子模拟:盘问卵白质、DNA瓜分子的折叠、构象变化和与药物分子的互相作用。
材料力学性能:模拟材料在拉伸、压缩、剪切等载荷下的应力–应变动作,盘问裂纹扩张和塑性变形。
热力学与输运性质:策划材料的热导率、粘度、扩散统统等。
相变经过:模拟液体结晶、玻璃调度、融化等经过。
值得肃穆的是,为了连结二者的优点,盘问东说念主员发展了多门径模拟要领。举例,第一性旨趣分子能源学(ab initio MD,AIMD)在MD的每一步齐使用DFT来策划原子间的力,从而在能源学模拟中已毕了量子精度,但其策划老本极高,仅适用于小系统和短时分模拟。
比年来,基于机器学习(ML)的势函数拓荒也成为贯串DFT和MD的紧要桥梁,它旨在用机器学习模子拟合DFT的策划驱逐,以达到接近DFT的精度和接近MD的策划速率。
小结
DFT和MD是策划模拟领域的两大利器,但它们基于弥散不同的物理旨趣。DFT是精准但竭力于的“显微镜”,潜入到量子层面揭示电子动作和化学实践;MD则是高效但通常的“录像机”,用于捕捉大门径、万古分的原子集体动态演化经过。
二者并非互相替代,而是功能互补。在科研施行中加拿大PC中国官网入口,应左证具体的科常识题和策划资源,采选最安妥的要领或将二者连结使用。